

Prof. Ehud Pines

Ben -Gurion University of the Negev , 1992-
Tel: 972-8-646-1572
Fax: 972-8-647-2943
epines@bgu.ac.il
Mailing Adress:
Chemistry department
Ben Gurion University
P.O.Box 653,
Beer-Sheva 84105, Israel
LATEST RESEARCH

Oxygen K-edge spectroscopy of hydrated proton complexes in solution provides compelling evidence of the pronounced impact on the electronic structure of the water molecules participating in the hydration, with a hierarchy in coupling strengths of the proton with profoundly strong orbital interactions for the inner three water molecules, and Coulombic field effects of the proton positive charge with the first hydration shell water molecules.

Photoacids have been understood, since the pioneering work of Theodor Förster more than seven decades ago, to show a pronounced increase in acidity when electronically excited, but the underlying microscopic mechanisms have remained elusive. Sebastian Eckert et al. show in their Research Article how time-resolved nitrogen K-edge spectroscopy along the Förster cycle of a photoacid provides key insight into the electronic structural factors responsible for the photoacidity phenomenon.

Spectroscopic fingerprints: We identify H7+O3 as the core protonated-water unit in water/acetonitrile solutions using IR and NMR spectroscopies. QM/MM MD simulations reinforce the experimental assignment and reveal the asymmetric structure of solvated H7+O3: a protonated water-dimer strongly H-bonded to a 3rd water molecule in a chain arrangement around the H7+O3 unit. Translocation of the H7+O3 motif driven by 1st shell H-bonds fluctuations around the H7+O3 unit lends itself to promoting the Grotthuss mechanism.

A “super” photobase experiences a 14-unit jump in pKa from its ground to excited state as the result of photon absorption. B. Borhan, M. Dantus, and co-workers report the discovery of a modular fluorenyl photobase capable of proton abstraction from organic solvents, unveiling the ultrafast dynamics of its intermolecular excited-state proton transfer process.
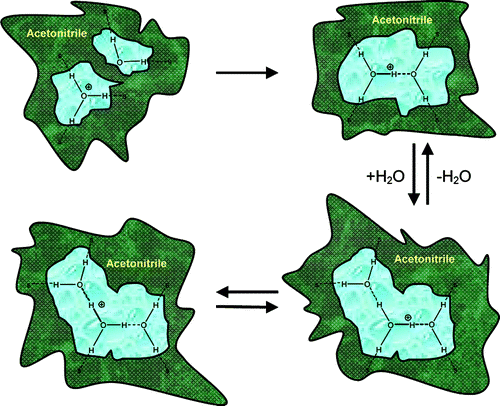
Gradual solvation of protons by water is observed by mixing strong mineral acids with various amounts of water in acetonitrile solutions, a process which promotes rapid dissociation of the acids in these solutions. The stoichiometry of the reaction XH+ + n(H2O) = X + (H2O)nH+ was studied for strong mineral acids. We have found by direct quantitative analysis preference of n = 2 over n = 1. the spectral features observed for n = 2, H5+O2, remain almost unchanged at large n values up to bulk water.

Bifunctional photoacids such as 6-Hydroxy-2-naphthoic acid and its sulfonate derivatives exhibit proton transfer from the acidic ro the acid group.In addition, change in the ionization state of one functional group causes a change (switch) in the reactivity of the other functional group. Using picosecond time-resolved and steady state spectroscopy, we find clear evidence for an ultrafast reactivity switch caused by a diffusional proton transfer through the water solvent between the two functional groups with no evidence of a concerted proton transfer.
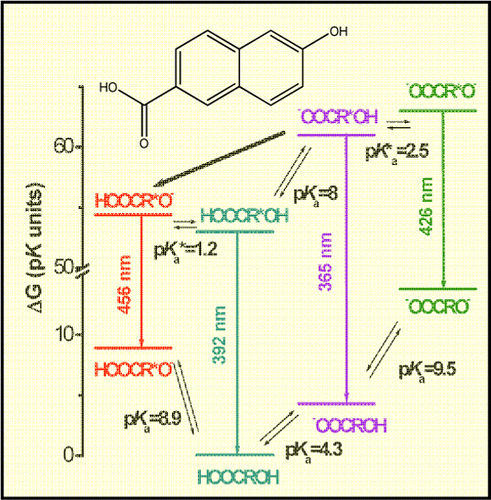
Reversible protonation (deprotonation) of a side-group is a useful and convenient way to affect the reactivity of large organic and biological molecules. We use bifunctional photoacids to demonstrate how the protonation state of a basic side-group (COO–) controls the reactivity of the main acidic group of the photoacid (OH), both in the ground and the electronic excited state of 6-carboxy derivatives of 2-naphthol.

Carbonic, lactic, and pyruvic acids have been generated in aqueous solution by the transient protonation of their corresponding conjugate bases by a tailor-made photoacid, the 6-hydroxy-1-sulfonate pyrene sodium salt molecule. The on-contact proton transfer (PT) reaction rate from the optically excited photoacid to the carboxylic bases was derived, with unprecedented precision, from time-correlated single-photon-counting measurements of the fluorescence lifetime of the photoacid in the presence of the proton acceptors. The on-contact PT rates were found to follow the acidity order of the carboxylic acids: the stronger was the acid, the slower was the PT reaction to its conjugate base. The pKa of carbonic acid was found to be 3.49 ± 0.05 using both the Marcus and Kiefer–Hynes free energy correlations. The considerable acid strength of intact carbonic acid indicates that it is an important protonation agent under physiological conditions.